This Behind the Scenes article was provided to LiveScience in partnership with the National Science Foundation.
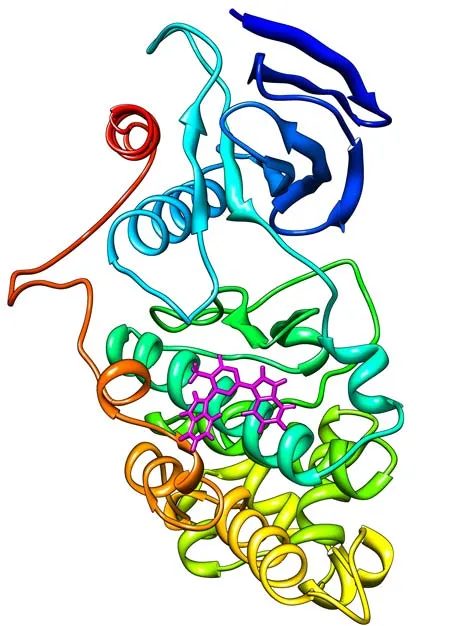
Most drugs enter our bodies as small molecules, ligands that bind to the surface of target proteins, inhibiting their function and protecting our health. For a drug to tame a headache or reduce a swollen knee, it needs to be effective at small doses, and selective enough to limit side effects.
With so many medicines to choose from on the shelves of your local pharmacy, it would seem that finding a new drug is a simple, straightforward process. In reality, discovering a new medicine can be a Herculean effort.
Latest Videos From
On average, it takes 15 years and more than $800 million in research and development for a drug to come to market, according to experts. This drives up the price of blockbuster drugs, while limiting research into less profitable medications.
Due to the time and costs involved, advanced computing is crucial to drug discovery efforts. By simulating the binding of virtual proteins and ligands, chemists can screen vast pools of possible compounds faster than would ever be possible in the laboratory.
This process trims the number of possible cures from millions to hundreds, at which point the drug candidates can be studied in the lab, thereby making drug discovery cheaper and faster. Several important HIV protease inhibitors were discovered using this method.
However, virtual "enrichment" is only helpful if the most effective molecules end up in the top 10 percent of the prediction. More often than not, they don’t, leading to frustration and skepticism in the field.
Pengyu Ren, assistant professor of biomedical engineering at The University of Texas at Austin, is trying to solve this problem. Using the NSF-funded Ranger supercomputer and a large pool of known protein-ligand matches, he is fashioning a robust way to search for new drugs.
"We're testing and developing computational approaches that can best reproduce the experimental data of protein-ligand binding that has been reported in the [academic] literature," Ren explained.
The effort is one of the most comprehensive studies ever undertaken of protein-ligand interactions using all-atom simulations. By representing physical reality with far greater fidelity, Ren expects the new method to significantly improve the effectiveness of drug discovery.
"In the old days, shortcuts were necessary to achieve speed. Researchers made approximations of physical models because the computations were too expensive," Ren said. "We're adding that further layer of physics to get more accurate predictions."
The physical reality of these simulations is enabled by supercomputers at the Texas Advanced Computing Center (TACC). "Virtual drug simulations require massive computing power," Ren said, "and by having access to TACC, we're in a position to actually try out these methods."
Ren is evaluating the accuracy and efficiency of different methods by applying them to more than 200 complexes across 10 different protein families. The method with the closest correlation to the real results is deemed the most effective. If the most successful method is proven to work consistently, Ren believes chemists will adopt it.
"The promise of rapid, inexpensive computational drug discovery has thus far eluded scientists," said Michael Gonzales, life sciences program director at TACC. "Pengyu's work is an excellent example of how current advances in computing power are enabling scientists to take a fundamentally different approach to virtual drug discovery."
It's not just about methods and protocols for Ren. He is also involved in a number of collaborations that put his algorithms to the test, exploring the relationship between rigidity and protein-ligand binding, and searching for inhibitors to proteins that are involved in cancer and other diseases.
"If this works, it will improve our ability to design drug candidates that are more potent with fewer side-effects," Ren said. "But to make robust, accurate predictions, it's time to invest in the next generation of computational technologies for drug discovery."
Editor's Note: This research was supported by the National Science Foundation (NSF), the federal agency charged with funding basic research and education across all fields of science and engineering. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author and do not necessarily reflect the views of the National Science Foundation. See the Behind the Scenes Archive.

 Live Science Plus
Live Science Plus